
Replication timing by comparative hybridization
Raghu's protocol; see Friedman, K. L., M. K. Raghuraman, W. L. Fangman, and B. J. Brewer (1995) Analysis of the temporal program of replication initiation in yeast chromosomes. J. Cell Sci. Suppl. 19:51-58.
The principle: A non-replicating DNA circle is created in vivo by site-specific pop-out recombination. This non-replicating fragment is then used as an internal control to measure the two-fold ncrease in copy number of replicating chromosomal sequences between the start and end of S phase.
| 1. | Grow the cells in minimal medium with 1% Na-acetate or 3% glycerol as the carbon source, aiming for about 20 ml cells per time point. (Raffinose has not worked well for me, so I avoid it.) |
| 2. | Arrest the culture with alpha factor (200 nM final concentration for bar1 strains) when the OD660 is about 0.2-0.25. |
| 3. | When the cells have arrested (more than 95% unbudded), remove 20 ml for the "Uninduced control", and add dry galactose (2% w/v final concentration) to the remainder of the culture. |
| 4. | Allow the induction to proceed for 2.5 hr (R-KT-DIR) or 4 hr (R-Zeo-DIR), then add dry glucose (2% w/v final concentration). Thirty minutes later, shift the culture to the 37° bath. |
| 5. | Once the culture has reached 37°C
(it takes 10-15 min), add Pronase (0.01-0.05 mg/ml final conc) to release
the cells from the alpha factor block.
[You can add dry Pronase. I usually dissolve the Pronase in 5 ml of minimal medium, just to avoid having the dry Pronase stick to the sides of the flask.] |
| 6. | Hold at 37°C for 90-120 min (until budded cells accumulate). Remove one sample (t = 0), then swirl the flask in ice-water and return it to 23°C. Continue to collect samples every 4 min (until t = 80 min or more). I usually take two 0-min samples, and take samples every 4 min from t=8 through t=56, then take t=64, 72, and 80 min samples--19 samples in all. |
| The ice-water treatment is to quick-chill the culture to 23°C, and is usually for ~1 min. You can empirically determine how long it takes to bring your desired volume of medium from 37° to 23°C. |
Collecting samples: Freeze 8 ml of 0.1% NaN3, 0.2 M EDTA in "50 ml" Oakridge-type plastic, screw-capped centrifuge tubes (frozen slanted to maximize the surface area). During the experiment, mix 20 ml of cells with 1/50 volume of 10% NaN3 in a 35 ml Corex tube (on ice) and immediately transfer the mix to a tube of frozen EDTA/NaN3. Alternatively, squirt the 10% azide on the frozen azide/EDTA and immediately add the cell sample. Vortex or shake the tube vigorously to chill the sample and break up the frozen EDTA. Spin down the cells in a chilled centrifuge, wash with 1 ml cold water per sample in Eppendorf tubes and freeze the pellets at -20oC.
| 7. | Extract the DNA (smash-&-grab with glass beads); digest 1/4 to 1/3 of it with the enzyme of choice in 40-60 microliters (EcoRI works well, as it cuts the ARS-less circle just once). Split each digest in two or three to run in duplicate (or triplicate) gels. It helps to make wide, well-separated wells. Blot the gels as usual. |
| 8. | Hybridize simultaneously with probes for the ARS-less circle, an early marker, a late marker, and the sequence of interest. Quantitation comes out best if the fragments to be probed are in the 2 kb-8kb range. Sometimes the fragments just don't work out (i.e., two fragments are too close for comfort), in which case you'll have to strip the blots and re-probe. If so, I usually include the ARS-less circle probe for each hybridization. |
| 9. | Quantitation: we use a PhosphorImager
or a Packard InstantImager. The values obtained for the two t=0 samples
are combined and used as the zero time point (to be used for normalization
of the later samples). Normalize each S phase sample against the t=0
controls:
Do the quantitation for each gel and plot the mean values for each time point. Here's a typical plot showing replication of ARS1 and R14 (a fragment near the right end of chromosome V):
|
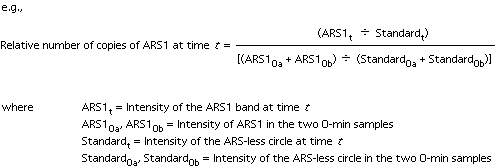
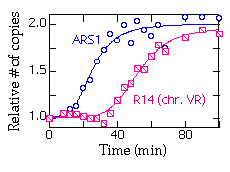
Strains: R-KT-DIR and R-Zeo-DIR are both derived from RM14-3a (MATa, cdc7 his6 leu2 trp1 ura3 bar1). The ARS-less cassette in R-KT-DIR is a Kanr-TRP1 fragment integrated near the right end of chromosome V. The t1/2for excision of this cassette is about 1 hr. The ARS-less cassette in R-Zeo-DIR contains the Zeocin-resistance marker and a portion of the bacterial Kanamycin resistance gene, and is integrated at LEU2. The t1/2 for exision of this cassette is about 2 hr at 23°C. Both strains have the Zygosaccharomyces rouxii "R" recombinase gene behind the GAL promoter integrated at LEU2. Both strains are Leu+.
Pronase: We use Calbiochem's Pronase (catalog #53702), which seems pretty clean--i.e., plates spread after Pronase treatment aren't contaminated--but I heard from someone who'd used cheaper Pronase from a different source and had massive growth of Streptomyces.